I've had the pleasure of corresponding with Yuhong Liang (梁雨虹)of 4A Biotech in China; she is the product manager for Morpholino oligos at 4A Biotech. We have been working with 4A for a little over a year now and Yuhong has been learning very quickly, but is still new enough to the Morpholino field that she is asking questions of broad interest. She had some questions regarding which splice junction to target, predicting cryptic splice sites, and the process of doing the two-non-overlapping oligo specificity experiment. I am trying to help her understand Morpholino techniques so she can help users directly in China, so I responded at some length. I'll post my response to her (with her kind permission) in hopes other Morpholino users might glean a useful idea.
Yuhong was looking at a specific transcript with three exons, having a start in exon 1 and a stop in exon 3.
--------------------------------------------------------------
"As the most common outcome is exon skipping, we should consider i1e2 and e2i2 first, right?"
When targeting the first and last splice junctions, oligos usually cause intron inclusion. This sometimes provides a strong gene knockdown, so in some cases these will be very good choices (see the third figure and its discussion on this page: Splicing outcomes: targeting for exon skipping or intron inclusion). However, because removing exon 2 will frameshift downstream sequence it is also a very good choice; my favorite starting place is to skip the most upstream exon available that will cause a frameshift.
"What's the difference between i1e2 and e2i2?"
The i1e2 is a splice acceptor site and the e2i2 is a splice donor site. I generally prefer splice donor targets because there is some extra sequence conservation in the intron of the splice acceptor site which increases the chance of an off-target interaction. Acceptor sites have a polypyrimidine region and a nucleophilic A base that are involved in binding snRNPs and closing the splice lariat. All splice junctions have a few very conserved intronic bases; introns start with "gu" at the donor site and end with "ag" at the acceptor site. The additional polypyrimidine region and nucleophilic A base at the splice acceptor decrease the possible base sequences that an acceptor site can have, so there is some similarity between all of the acceptor sites in the transcriptome. That means the chance of an acceptor-targeted Morpholino binding to an unintended acceptor splice site is greater than the chance that a donor-targeted Morpholino will bind to an unintended donor splice site (because the donor sites can have more sequence variation). When someone asks for a Morpholino targeting a particular exon, I will try first to design a splice donor oligo and then if the oligo characteristics do not look good I will see if there is a better-looking splice acceptor. You can produce a stronger effect by using both donor and acceptor site oligos together at the same time; they should produce dose synergy.
"How can we confirm if there is cryptic splice site in exon2 before PCR?"
I don't know of a method for checking if there is a splice site prior to PCR. You can check the sequence to see whether there are AG or GT sites away from the splice junctions and these could be cryptic splice sites, but I think you will see many AG and GT sites which do not participate in splicing. Even the canonical GU starting an intron and AG ending the intron is not perfectly conserved; while the major spliceosome requires these bases, the minor spliceosome can use GU or AU at the acceptor and AG or AC at the donor site. There are some other semiconserved residues at active splice sites, for instance at a splice donor there is commonly an AG at the end of an exon just before the GU in the intron, but the exonic AG bases are not perfectly conserved -- their presence or absence give a hint about a cryptic site but not certainty. Cryptic splice acceptor sites need the polypyrimidine and nucleophilic A residues to function, but the configuration of these bases (e.g. distance from the junction) can be shifted about a bit.
The first method for checking whether or not a cryptic splice site is redirecting splicing is to do PCR and run the PCR products on an electrophoretic gel, comparing PCR products from Morpholino-treated and untreated cells/embryos/animals. If it is an easy splice, like successful clean excision of an exon, one gel can usually provide all the information needed but, if something unexpected occurs during splicing, then it might take several primer sets to determine what happened. A tricky situation is if a splice is redirected to a very close cryptic splice site. This can cause a very slight change in the migration of the PCR product band on the gel. This kind of short-distance splice-redirection can be revealed by sequencing the RT-PCR product; if a lab can afford to do the sequencing, it provides the most clear assessment of a splice oligo outcome. You see exactly where the cryptic splice site lies and you can predict if a frameshift occurred or not after oligo treatment.
"Are there two primers as below?"

That is the set that I would start with, and if it shows the expected outcome then that set is probably enough. If there is an unexpected intron inclusion, it might take a primer targeted in the intron to show that the inclusion has occurred.
"If designing for a wild splice outcome, how many Morpholino oligos should be needed?"
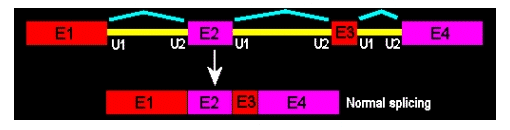
Wild-splice is the outcome when no Morpholino oligos are used. Think of "wild-type" vs. "mutant"; for splicing Morpholinos, we can talk of "wild-spliced" vs. "morphant" or “exon-skipped”. Here, no exon-skipping occurred; this is the mRNA as it is used by the cells to make functional proteins.
"About the two-nonoverlapping-oligos: should the oligos be used in separate experiments or by co-administration? When co-administrating, what concentration should be used?"
There are several possibilities for the non-overlapping oligo specificity control experiment.
First, try the oligos one-at-a-time, in separate experiments. If the morphants phenocopy each other, then that suggests that the oligos are specific and the observed phenotype is a result of interaction of the Morpholinos with their intended target; the probability is low that the same phenotype would be produced from both oligos due to interaction with an unintended RNA. However, there are RNA families that require additional concern; for instance, if a gene was duplicated and over many generations the RNA sequences from each duplicate began to diverge, but this happened recently enough that the sequence has not changed very much, then it is possible that two Morpholinos targeting a single RNA might both interact with the closely-related RNA produced by the gene duplication.
Next, you can try the two oligos together to see if there is dose-synergy. This is not done routinely, but is a good specificity experiment. Dose-synergy means that there is a greater-than-additive effect if the oligos are used together. If you get the same outcome using one of the oligos at some dose, let's call it one unit, then you get the same outcome if you use both oligos together each at 0.5 unit dose, then that does not show synergy, it is simply additive: 0.5U + 0.5U = 1U. However, if you use 0.25U of each oligo and you get the same outcome that you see from a single oligo at 1 U, that is an example of dose synergy: 0.25U + 0.25U < 1U (but caused the same outcome). Dose synergy is an indication that the oligos are interacting with the same mRNA (although it is still possible that they are interacting with two different RNAs in the same biochemical or physiological pathway). Dose synergy was proposed as a test for oligo specificity in this paper:
Bill BR, Petzold AM, Clark KJ, Schimmenti LA, Ekker SC. A primer for morpholino use in zebrafish. Zebrafish. 2009 Mar;6(1):69-77.
http://www.liebertonline.com/doi/pdfplus/10.1089/zeb.2008.0555
Base the initial doses for the co-administration dose-synergy experiment on a low dose of the single oligo that produces a phenotype. There is a factor that makes this more complicated, which is that different oligo sequences will have different effective dose ranges. Because of this, both oligos need to be tested individually to find their effective range, especially to determine the lower end of that effective range. Let's call those low effective doses U1 and U2 for oligo 1 and oligo 2. If you try a dose synergy experiment at 0.25 doses, you would use 0.25U1 of oligo 1 and 0.25 U2 of oligo 2. This corrects for the difference in activity of the two oligos.
You can also try the two oligos at 1 U of dose each, co-injected, to look for the effect of a stronger knockdown than is available with a single oligo.
There is a new sequence specificity experiment emerging, which uses a null-mutant embryo and a Morpholino targeting the RNA with the null-mutation; this has so far been explored in zebrafish embryos. If a Morpholino targeting a particular gene's RNA produces a measurable phenotype in a wild-type embryo, and then that Morpholino is injected into a mutant null for that gene and no change in phenotype is seen, this supports the hypothesis that the phenotype of the Morpholino in the wild-type is due to a specific interaction with the targeted RNA. If, however, the Morpholino still produces some phenotype in the null-mutant background, that does not preclude specificity; it is possible, for instance, that the Morpholino is interacting with maternally-deposited RNAs (this can be tested by injecting the Morpholino into embryos from a homozygous-null mother).
Sometimes a mutant does not produce the same phenotype as a wild-type embryo injected with a Morpholino targeting the gene that is null in the mutant. However, if that Morpholino sequence is injected into the null-mutant embryo then the Morpholino phenotype can disappear. That is the case if the mutant is compensating for the loss of the gene of interest by altering the expression of other genes; this is homeostasis, the tendency of a stressed organism to readjust its physiology to attain a healthy state. When that Morpholino is injected into the null mutant, it has no functional target (the target is the null) and other genes have adjusted their expression to compensate for the loss of the null gene, so you don't see the Morpholino phenotype. This was initially explored in this paper:
Rossi A, Kontarakis Z, Gerri C, Nolte H, Hölper S, Krüger M, Stainier DYR. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015 Aug 13;524(7564):230-3. doi: 10.1038/nature14580. Epub 2015 Jul 13.
http://www.nature.com/nature/journal/vaop/ncurrent/full/nature14580.html
I recently had a nice discussion about this oligo specificity control strategy with Martin Blum, which is on my blog here:
Validating Morpholino phenotypes with CRISPRs
